Medical Devices
FDA Busts Johnson & Johnson and Sientra Over Breast Implant Safety





Comments
Comments

Comments


Defective Products11 months ago
Firefighting Foam Lawsuits Surge as Health Risks Linked to “Forever Chemicals”

Negligence & Personal Injury1 year ago
Understanding Video Game Addiction in Children: A Growing Concern for Parents

Dangerous Medications2 years ago
Depo-Provera Birth Control and Brain Tumors: Current Evidence and Research Gaps


-

 Defective Products6 years ago
Defective Products6 years agoDehumidifier Recall: Several Brand Name Humidifiers are Recalled as a Fire Hazard
-
Nursing Home Abuse7 years ago
Hundreds of Allegations at Georgia Elder Care Facilities Being Investigated
-
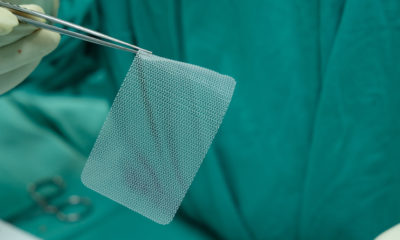
Medical Devices7 years ago
Complications from Hernia Mesh
-
Covid-196 years ago
Metformin as Potential Protector from COVID-19
-
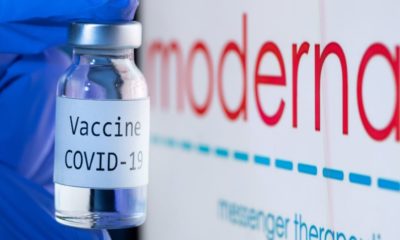
Covid-195 years ago
Moderna Seeks Emergency Approval for COVID Vaccine
-
Prescription Drugs5 years ago
Recall for Drug Mix-up with Serious Effects
-
Prescription Drugs6 years ago
Some Heart Meds May Come with Extra Risk
-
Defective Products5 years ago
Excedrin Issues Recall for Headache Medicine