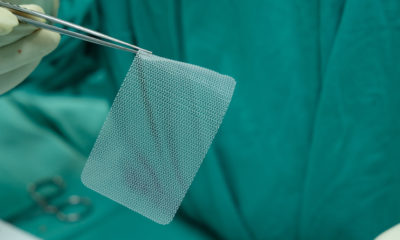
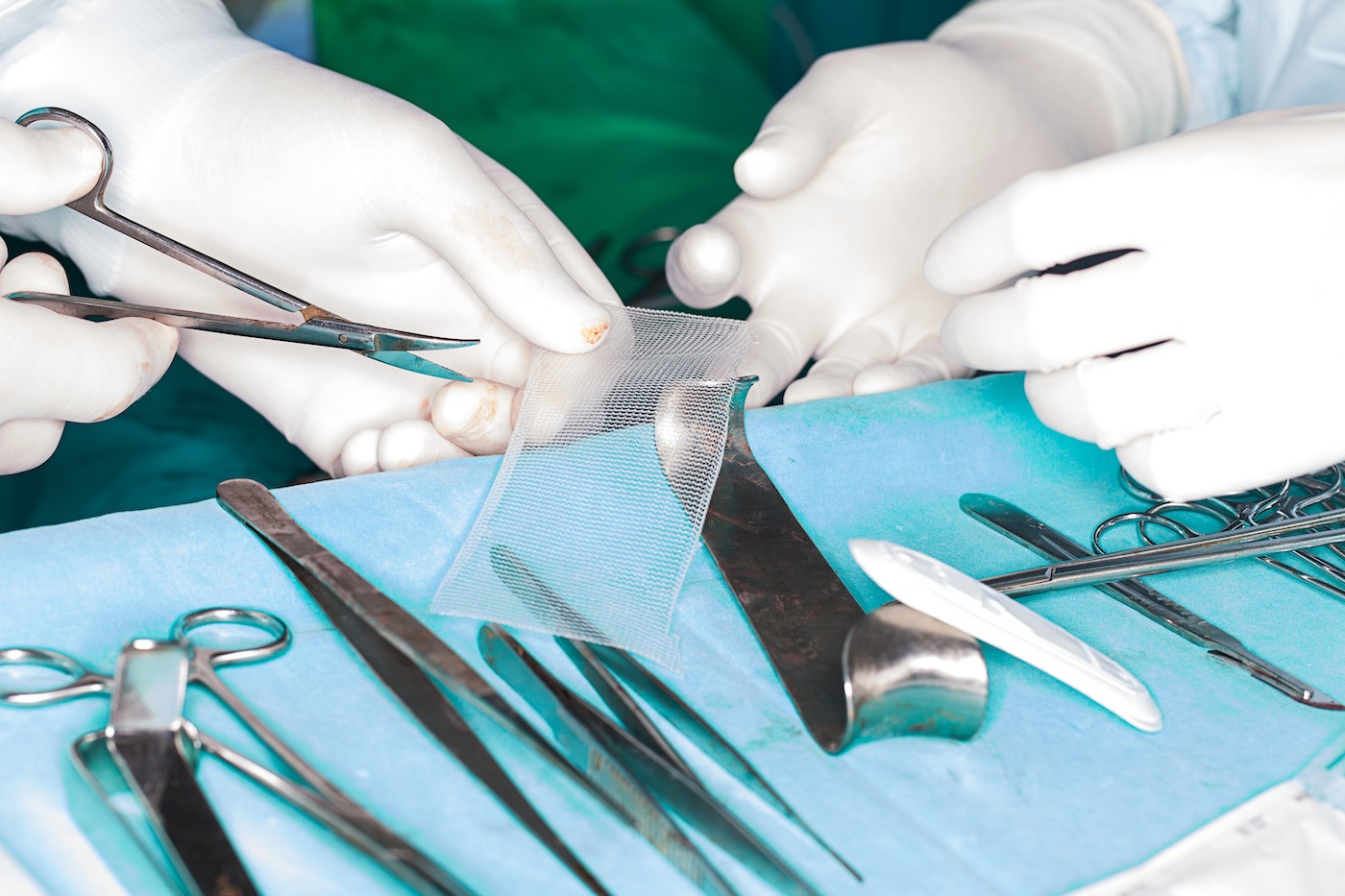
The U.S. Food and Drug Administration has just flagged a common medical device—surgical staplers and implantable staples—because of a heightened risk associated with their use in medical procedures.
The FDA issued its warning in a “Letter to Health Care Providers.” In it, the federal agency is alerting doctors and health care providers of an alarming increase in reports of malfunction when using surgical staplers and implantable surgical staplers. In its warning, the FDA also published recommendations for how manufacturers can improve the labeling of surgical staples and stapler devices to help health care providers use the devices more properly and effectively, such as choosing the correct size of staple for the type of procedure and patient tissue. The labeling recommendations also suggest that health care providers consider alternative options to surgical stapling in cases where the patient has a predisposition of bleeding or if the patient has swollen tissues.
The FDA also highlighted its own recommendations to help reduce the risks of these devices, and they outline the agency’s plans to address the safety of the devices over the coming months. These plans include creating a public advisory committee to analyze the path for medical device manufacturers to bring their surgical staples and staplers to market, and whether this current pathway has the appropriate safety and review checks.
Thousands of Reports of Device Malfunction.
The FDA’s warning reports that it has received more than 41,000 risk reports for surgical staplers and implantable staples since January 2011. These complaints, called “medical device reports,” include more than 90,000 serious injuries, more than 32,000 reports of device malfunction, and 366 patient deaths from using these devices.
The agency outlined the types of problems reported from surgical staplers and staples. The most common issues reported range from staplers misfiring and difficulty of firing staples, to malformation of staples and opening of the suture line after implantation. The complaints reported also included an issue with the staples being misapplied, or when the health care professional implants the staples into the incorrect tissue, as well as cases of using the wrong size of staple for the patient’s tissue.
The FDA states that these problems with the devices can have significant impacts on the health and safety of patients. Health risks include prolonged surgical procedures and risk of tearing of internal organs and tissues. The malfunction of surgical staplers and implantable staples have, in some cases, required additional, unplanned medical procedures; these, in turn, could lead to other medical complications including bleeding, tissue and organ tearing, increased risk of cancer, and even death.
And this is not the first time that the FDA has warned about the risks associated with surgical staplers and implantable staples. The FDA has been reviewing the risks of these devices for years and has issued previous communications about the potential risks of their use.
Prevalent Use of Surgical Staplers and Implantable Staples.
Surgical staplers and staples are widely used across many types of surgeries and medical procedures. These procedures range from gynecologic and gastrointestinal procedures, to procedures creating connections between organs and body tissues. Implantable staples and surgical staplers also facilitate resection procedures, or when surgeons remove part of a patient’s organ.
One benefit of surgical staplers and staples is a quicker operating time or the patient: the devices help make surgical procedure time quicker than it would be with traditional manual suturing. Implantable staples also have a lower risk of infection and maintain a strong wound closure to help support the patient’s recovery.
Quick Market Access for Device Manufacturers.
This warning letter from the FDA has highlighted the quick and easy access to the market that manufacturers of surgical staplers enjoy. Because these devices are categorized as only “Class I” medical devices, they do not require any regulatory submissions to the FDA before they can be sold on the market to health care professionals. This means that there is no mandatory FDA review and approval before these surgical staplers are used in medical procedures. (On the other hand, surgical staples are classified under the higher “Class II” of medical devices, and therefore they do require the FDA’s review before they can go to market.)
The Investigation is Ongoing.
In response to its new warning, the FDA emphasized that reporting potential health risks to the pubic is essential to the agency’s public health mission. The chief medical officer of the FDA Center for Devices and Radiological Health said that the agency also plans to make its own process of addressing medical device risks more efficient, modernizing its procedures so that it can address device safety issues more quickly. This includes sharing information and analysis of medical device benefits and risks to both the public and to health care providers.
The FDA also emphasized that it is a top priority of the agency to improve the safety of surgical staplers and implantable staples. Meanwhile, its investigation into surgical staplers and staples is ongoing.
















 Defective Products6 years ago
Defective Products6 years ago